AutoDock CrankPep v1.1
The older and deprecated version of ADCP (Version 1.0) is available here
AutoDock CrankPep is part of the AutoDock Suite.
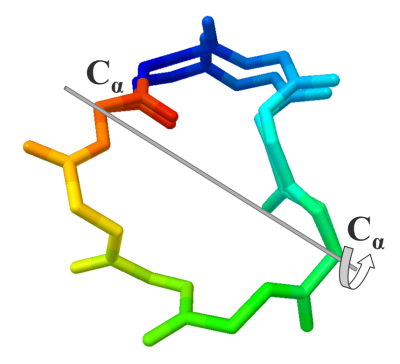
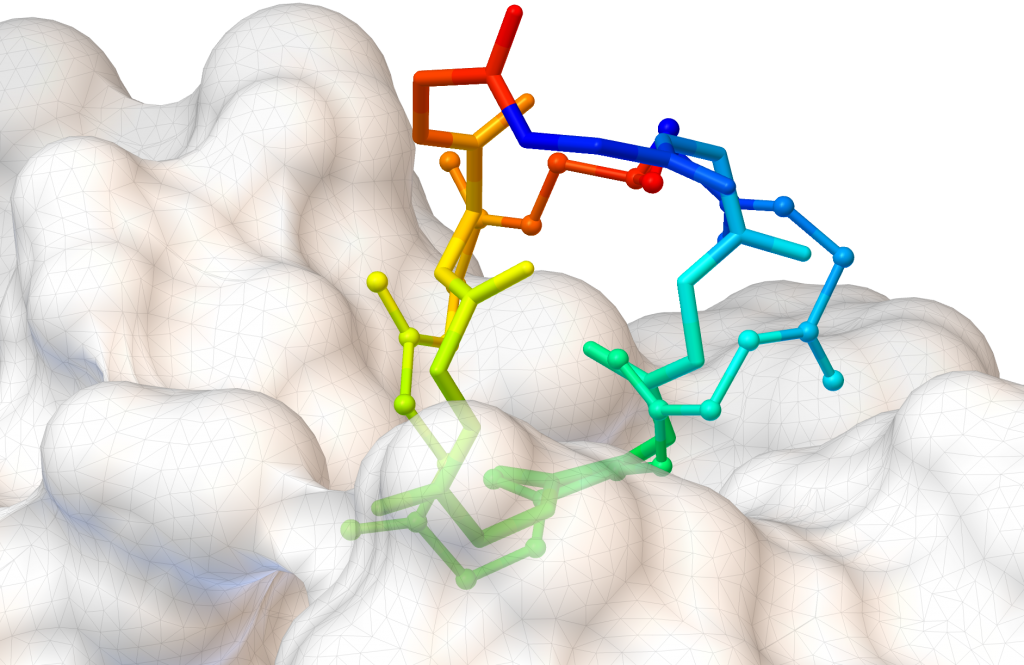
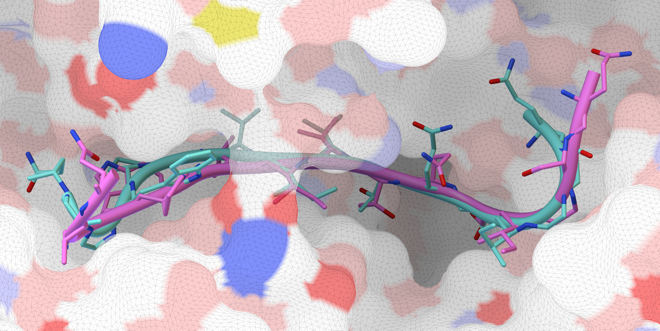
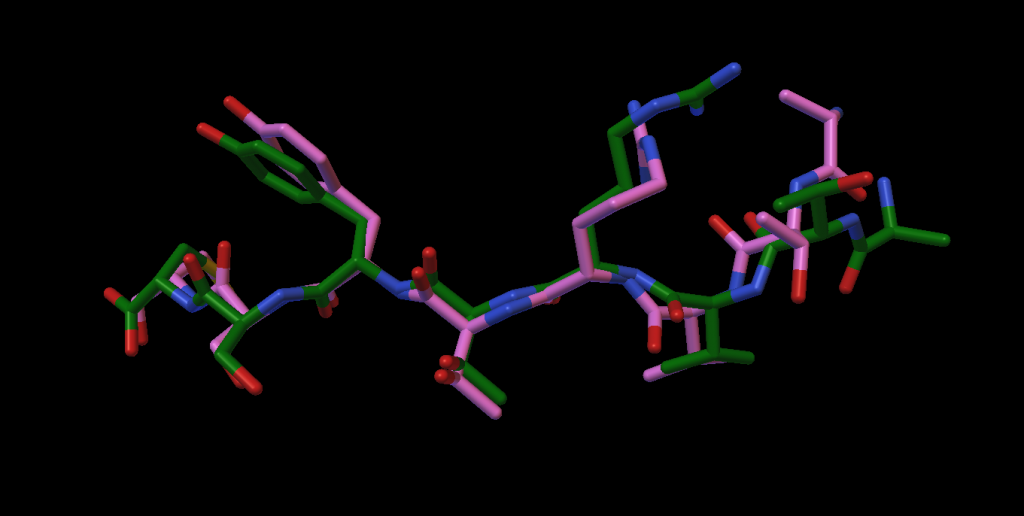
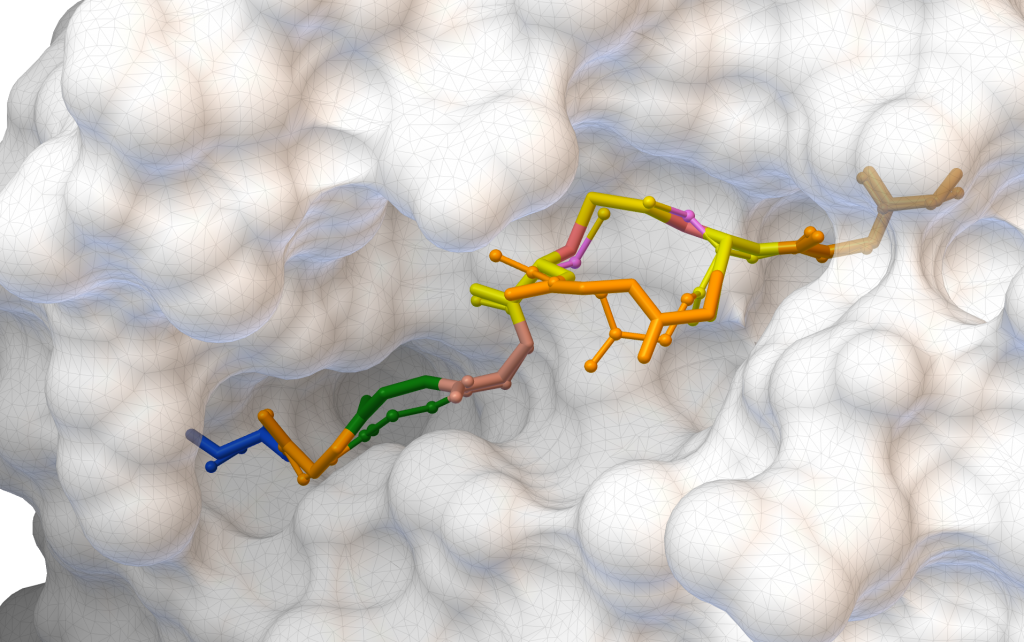
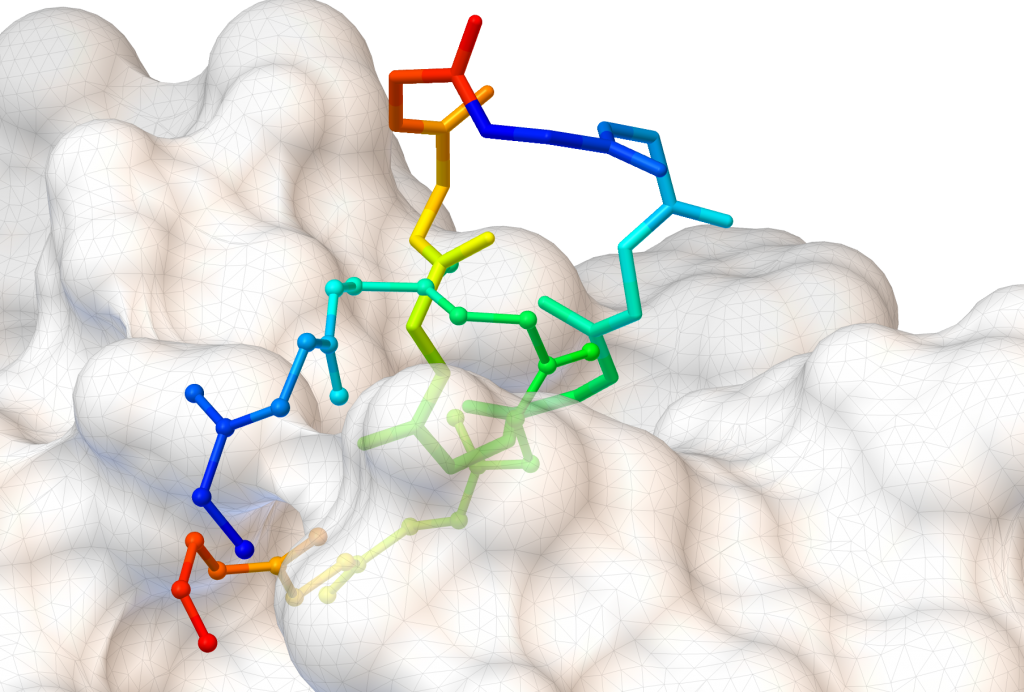
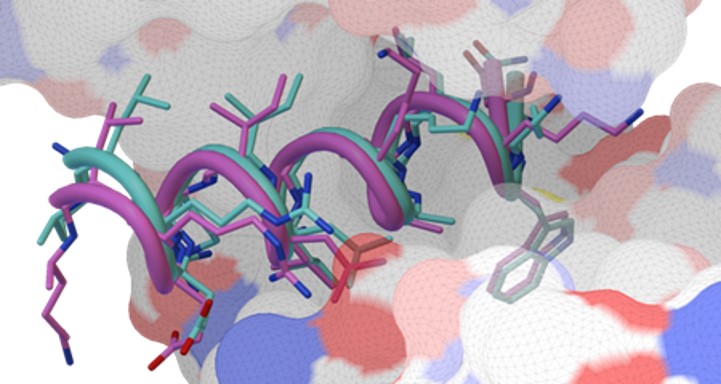
AutoDock CrankPep (ADCP) is a specialized docking engine for peptides that uses a protein folding technique along with AutoDock’s efficient rigid receptor representation. It utilizes a Monte-Carlo search to fold the peptide while optimizing its interaction with the receptor, resulting in docked peptides. ADCP can handle peptides provided as a sequence string and has shown success with peptides of up to 20 amino acids.
In version 1.1, several improvements have been made, including support for non-standard amino acids and the ability to minimize docked poses using OpenMM. The changes include:
- Streamlined output: Docked poses are now saved in a single multi-model PDB file named “xxx_out.pdb,” where “xxx” is the docking job name (or a default name) and a summary file named “xxx_summary.dlg” is generated.
- Improved representation: Docked poses now include all-atom side chain representations, rather than a rotamer index.
- User-defined number of docked poses: Users can set the number of docked poses using the “-m” or “–nmodes” command line option.
- Command line changes: The target file specification has changed to “-T,” and it can accept a .trg file or a folder containing a .trg file. A new option, “-k,” allows users to keep calculation details.
- Rotamer libraries: Rotamers for amino acid sidechains are loaded from text files with .lib extension, and system rotamer libraries can be specified using the “-L” option.
- Support for non-standard amino acids: Version 1.1 has been modified to facilitate adding support for non-standard amino acids, allowing for the addition of rotamers for modified sidechains without recompiling the code. The syntax “<FOO>” is used to specify a coarse potential for an amino acid and a different side chain for building the full atom representation.
- Support for D-amino acids: Chirality for D-amino acids is indicated by “&” before the coarse potential.
- Protonation states for histidine: Support for different histidine protonation states (HIE and HID) has been added.
- Automatic loading of affinity maps: ADCP now automatically loads affinity maps for all detected atom types in the peptide.
- MD refinement of docked poses: Docked poses can be minimized using OpenMM, with options to control this feature.